Serendipity and Science
On the origins of chemical species, by Dan Lednicer
The enormous strides that have recently
been made in molecular biology hold great promise for speeding the discovery of
pharmaceuticals to treat diseases that have so far been recalcitrant to drug
therapy. The day may well be in the offing when a majority of important new
pharmaceutical products will owe their existence to carefully crafted research
programs based on the increasingly detailed understanding of the molecular
biology involved in the particular disease that is being addressed. The cost of
drug discovery and development has become so high that research management is
eagerly looking forward to a more systematic and hopefully predictable process
than that which has prevailed in the past. The genealogy of quite recently introduced drugs however provides a
good illustration of the role that serendipity, intuition or even pure chance
have played in drug discovery up until quite recently.
 The chemical structures of these compounds
can be traced back to a drug first introduced over half a century ago.
Molecular manipulation by organic chemists over the next fifty years led to the
discovery of at least three compounds each of whose annual sales are currently
in the billion-dollar range. The activity of the first of these, the oncology
product, Novaldex (tamoxifen) can be extrapolated from that of the compound
that started the research. The biological activity of the other two
descendants, the selective NSAIDs Celebrex (celecoxib) and Vioxx (rofecoxib)
could hardly have been predicted.
The chemical structures of these compounds
can be traced back to a drug first introduced over half a century ago.
Molecular manipulation by organic chemists over the next fifty years led to the
discovery of at least three compounds each of whose annual sales are currently
in the billion-dollar range. The activity of the first of these, the oncology
product, Novaldex (tamoxifen) can be extrapolated from that of the compound
that started the research. The biological activity of the other two
descendants, the selective NSAIDs Celebrex (celecoxib) and Vioxx (rofecoxib)
could hardly have been predicted.
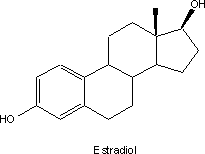
The story begins in the 1930s with the
discovery of the estrogens. The state of structural determination was still in
its infancy; the various instrumental methods now used routinely to unravel
structures had not even been conceived. By dint of hard and very elegant work
the structure of principal hormonal agent, estradiol, was found to be a based
on the so-called steroid nucleus.
It was known by then that low levels of
estrogen were associated with some diseases suffered by women. A promising
approach for treating those conditions involved increasing hormone levels by
administering pure estradiol. Supplies were however a major problem since the
compound occurred at such low levels that it was not considered practical to
isolate it from animal sources. (It is interesting, as an aside, to note that
exactly such an extract, Premarin, which consists of conjugated estrogens
isolated from mare’s urine comprises the enormously successful drug used mainly
by postmenopausal women).The methods
available in the 1930’s for chemical synthesis were not yet up to the task for
preparing by total synthesis what were then considered to be complex
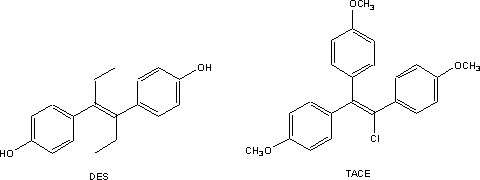
structures. The search for purely synthetic structurally simpler compounds that
showed estradiol-like activity led to the discovery of the ill-fated drug,
diethyl stilbestrol (DES). Subsequent research was to show that the receptor
for estradiol is notoriously promiscuous and will interact with a large variety
of compounds that only vaguely resemble its endogenous activator. Omitting one
of the ethyl groups and replacing the other by a substituted benzene ring led
the yet another purely synthetic compound, chlorotrianisene (TACE) that was
used as an estrogen in the early 1940s.
Both DES and TACE are poorly soluble in
water making it difficult to administer those drugs by injection. The phenolic
groups in DES are too weakly acidic to ionize at physiological pH while TACE
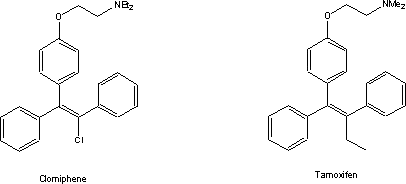
lacks any ionizable groups whatever. Arguably prompted by this chemists at the
Richardson Merell Company tackled the problem in the late 1950s by attaching an
amino group that would form salts with acids to one of the benzene ring of a
TACE-like molecule by way of a so-called basic ether1. The resulting
products were, as hoped, somewhat more soluble in water than the parent
compounds. To their surprise however those basic ether derivatives were no
longer simple estrogens. Instead they also showed a measure of anti-estrogenic
activity. In addition, because of that new activity many also acted as a
contraceptive in the rodent model then in use for screening compounds for
antifertility activity. Further refinement of the structure led the company to
market one of these drugs as clomiphene. This compound, which is a mixture of
cis-trans isomers, shows a mixture of estrogenic and antiestrogenic
activities.
Reports of this discovery occasioned the
start of work in a number of other pharmaceutical laboratories aimed at
exploiting this unforeseen lead. Chemist at the then ICI Company (now part of
Aventis via Zeneca) replaced the potentially unstable chlorine by an ethyl
group. They then investigated the resulting compound, tamoxifen, extensively as
an estrogen antagonist in the 1960s. The growth neoplasms such as breast
cancers were known even then to be stimulated by circulating estrogens. Heroic
measures, such as hysterectomy or administration of androgenic drugs, had been
used by oncologists of the day to treat breast cancer. Clinical trials showed that much the same
effect could be accomplished with oral doses of tamoxifen. The concurrent
development of tests for estrogen sensitivity of tumors allowed clinicians to
select those patients that would profit from drug treatment. Tamoxifen today
comprises mainline adjunct post-operative therapy for estrogen receptor
positive breast cancer2.
Several other groups on the other hand
focused on the antifertility activity displayed by the Merell compounds hoping
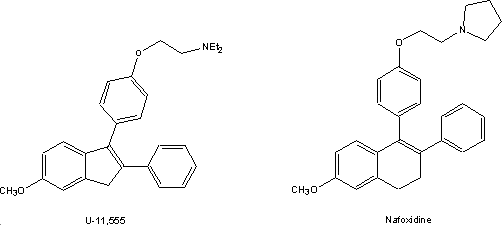
to find non-steroidal contraceptives. Chemists Upjohn designed an analogue that
included a new ring. This would, they thought, more closely resemble a steroid.
The new ring was five-membered since this was accessible in a relatively few
steps. The fact that these compounds were active in the screen led to a full
scale analogue program. The most potent indene of the series,U-11,555A3
went as far as Phase I clinical testing; it failed at this point because it
caused photosensitivity.
The ring expanded dihydronaphthalenes that
were somewhat more difficult to prepare turned out to be uniformly more potent
than corresponding indenes4. One of those, U-11,000A, replaced
U-11,555A in the clinic. Though the compound failed as a contraceptive it was
tested successfully by the National Cancer Institute as an estrogen antagonist
under the USAN name nafoxidine.
Some years later, the series was revisited
by scientists at Lilly. The structures of the compounds that they investigated
differed from those prepared at Upjohn mainly by the inclusion of a ketone
carbon atom (C=O) between the fused ring and that which holds the side chain
with the amino group. This will of course subtly alter the shape of the
molecules. Increasing sophistication of pharmacological tests had led to the
identification of subtypes among receptors, including those that recognized
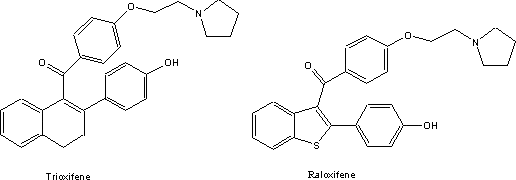
estrogens. These two compounds, trioxifene5 and raloxifene6
were investigated at in some detail because their pattern of activity on the
receptor subtypes differed from the classical estrogens and their antagonists.
The pharmacological response pattern of these compounds was deemed to be
particularly suitable for alleviating menopausal and post-menopausal symptoms.
This selective activity has led the Lilly scientists to refer to them as 'designer estrogens'. These compounds, like many estrogens are in addition
quite useful in treating or preventing osteoporosis. Raloxifene is currently
used extensively under the trade name of Evista.
The activities of the compounds that have
been considered up to now result from interaction with a receptor that
recognizes estradiol, the agent that prompted the series to begin with. The second half of the story is more
interesting in that it involves a totally unrelated biological target.
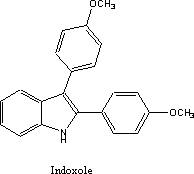
Another group at Upjohn was heavily involved
in indole chemistry at roughly the same time as the work on the indenes. They
prepared a series of indoles that carried two benzene rings in the same
position as those on the indene U-11,555. One of these unexpectedly showed very
good anti-inflammatory activity. This compound was taken to the clinic under
the generic name indoxole. This agent like its indene predecessor also failed
Phase I testing; interestingly for the same reasons. It too caused intolerable
photosensitivity.
There was great reluctance to walk away
from this lead because of the very good activity the compound had shown in
animal model. The fact that it was not related to any of the then-known
anti-inflammatory agents added reason to try to salvage this lead. The major
program aimed at producing non-phototoxic analogues of indoxole gave
disappointing results. Most of the modified indoxoles lost anti-inflammatory
activity; the few analogues that did retain activity also retained the
toxicity. It became quite clear that the phototoxicity in both this molecule
and its precursor, U-11,555 was due to the shared structural feature: the
presence of two benzene rings on the double bond in the fused five-membered
ring7.
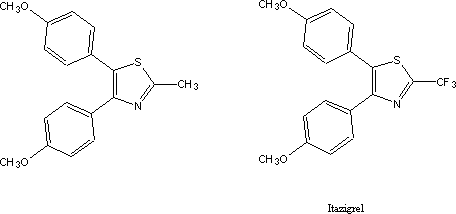
A modest series of analogues was prepared
to find out whether the indole ring could be replaced by a simpler five
heterocyclic ring. The two methoxy-substituted benzene rings were retained
since the earlier program had indicated that they were crucial for anti-inflammatory
activity. One of those, the dianisyl thiazole below, interestingly, retained
anti-inflammatory activity. Though the level of activity was too low to prompt
further follow-up it was however good enough to prompt the filing for a patent
on this and some of its derivatives8.
The indoxole program dates back to the
mid-1960s, just at the time that Upjohn was becoming ever more deeply involved
in prostaglandin research. The biochemical role of these hormone-like
substances was still far from clear. The discovery that prostaglandins were
directly involved in such injurious responses as inflammation and platelet
aggregation that leads to thrombosis and strokes was still some years off. By
the mid-1970s however Vane and his colleagues had demonstrated that nonsteroid
anti-inflammatory drugs (NSAID) worked by inhibiting an enzyme – cyclooxygenase
– involved in the biosynthesis of prostaglandins. Inhibitors of that enzyme
will also counteract platelet aggregation. A new screen for such inhibitors
picked up the old thiazole lead. The ensuing analogue program culminated in the
synthesis of a compound in which the hydrogens on the methyl group were
replaced by fluorine9. This
agent showed enough activity so that it was assigned a generic name, itazigrel,
an indication that it was a clinical candidate. Publications as recent as 1994
identify itazigrel as an inhibitor of cyclooxygenase10.
Research on new and improved NSAIDs had
pretty much run its course by the mid-1990s. There is room on the market for
only so many 'profens'. All these drugs seemed to have the same propensity for
contributing to stomach ulcers. By then it had been fairly well established
that this side effect was in fact a consequence of the drug’s action on
cyclooxygenase. This is due to the fact
that the enzyme is also involved in the synthesis of prostaglandins that
normally protect the lining of the stomach. . The discovery that the enzyme
occurs in two forms led to a new wave of research on NSAIDs. One of those
enzymes, COX-2, seemed to be involved in inflammation but not in the chain of
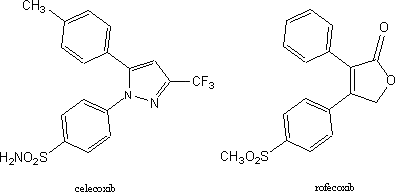
events that led to ulcers. In 1998, G.D. Searle, by then part of Monsanto,
announced that they had developed a specific COX-2 inhibitor. This drug
Celebrex (celecoxib) was heralded as an NSAID with much reduced effects on the
stomach. Examination of the structure of this compound shows that it bears an
interesting relation to itazigrel and its ancient predecessor. The relation becomes even closer when one
considers that one of the methoxyl groups (CH3O) in itazigrel is
almost certainly cleaved to a phenolic hydroxyl (OH) in the body. It is well
known that sulfonamide groups (H2NO2S) such as that
present in celecoxib are often biologically equivalent to phenols. The structure of newer COX-2 drug,
refecoxib, still retains the basic backbone though with many changes in
functionality.
References:
1. R.E. Allen, F.P. Palapoli, E.L.
Schumann, M.G. Vancampen, US Patent 2,914,563 (1959).
2. G.R. Bedford, D.N. Richardson, Nature, 212, 733(1966).
3. D. Lednicer, J.C.
Babcock, P.E. Marlatt, S.C. Lyster, G.W. Duncan, J.Med.Chem., 8, 52(1965).
4. D. Lednicer, S.C.
Lyster, B.D. Aspergren, G.W. Duncan,
J.Med.Chem., 9, 172(1966).
5.
C.D. Jones, T. Suarez, E.H. Massey, L.J. Black. F.C. Tinsley, J.Med.Chem., 22, 962(1979)
6. C.D. Jones, M.D.
Jevnikar, A.J. Pike, L.J. Black, A.R. Thompson J.F. Falcone, J.A. Clemens,
J.Med.Chem., 27,
1057(1984)
7. J. Szmuszkovicz, E.M.
Glenn, R.V. Heinzelman, J.B. Hester, and G.A. Youngdale, J.Med.Chem., 9, 527(1966).
8. D. Lednicer, U.S. Patent 3,560,514
(1971).
9. R.H. Rynbrandt, E.E. Nishizawa, D.P. Balgoyen, A.R. Mendoza, K.A.
Annis, J.Med.Chem.,24,
1507(1981).
10. A. Tanaka, H. Sakai, Y. Motoyama, T. Ishikawa, H. Takasugi,
J Med Chem., 37,1189(1994).
All opinions expressed in this article are those of the author and are not necessarily those held by DBSW,
sciencebase.com or its associates.