Brainstorming chemistry
by David Bradley
ChemBrain doesn't look unusual. Straightforward Windows interface and dialog boxes, albeit, old style and slightly inelegant in places. But, aesthetics aside, ChemBrain is different. There are numerous chemical databases, spreadsheets and simulation packages that have, in recent years, provided inbuilt chemical awareness. This savoir faire allows them to recognise a structure for what it is rather than simply "seeing" a cluster of lines and letters as is the case with a conventional, non-chemical drawing package.
 But, ChemBrain seems to take this inherent knowledge in a different direction. This unique chemical database for three-dimensional molecular structures brings with it integrated artificial intelligence. ChemBrain's AI capabilities allow it to learn about molecules so that not only is structure understood by the program but it can help users predict almost any molecular property for that structure. Add to that the fact that it works well as an electronic lab-book and what more could you want from a chemistry program?
But, ChemBrain seems to take this inherent knowledge in a different direction. This unique chemical database for three-dimensional molecular structures brings with it integrated artificial intelligence. ChemBrain's AI capabilities allow it to learn about molecules so that not only is structure understood by the program but it can help users predict almost any molecular property for that structure. Add to that the fact that it works well as an electronic lab-book and what more could you want from a chemistry program?
As I said earlier, ChemBrain is fairly conventional in appearance, simple to use and allows quick and easy input of three-dimensional molecules. Click a bond type and start sketching. The program's geometry-optimizer cleans up the bond angles and lengths as you draw. Unfortunately, it's easy to get carried away and only with a little practice will the inadequate single-level "undo" stop being such a frustration. However, there are numerous example molecules included with the program and any number can be imported in the well-known MDL mol file format. A force-field algorithm conveniently converts the molecule into an acceptable 3D version.
One problem for molecular importers, which is admitted to in the help file, is that each item has to be defined, generic R groups and non-explicit halogens, marked X, will prevent such a molecule from being imported.
 Usefully, ChemBrain always checks whether a newly drawn chemical structure is already stored in its database. The search is not based the name of the molecule but on the structure so inadvertent double storage is prevented (although optionally possible for different conformations). This fact can even be used to find the name of a structure if only the structure is known (provided the compound is in the database).
Usefully, ChemBrain always checks whether a newly drawn chemical structure is already stored in its database. The search is not based the name of the molecule but on the structure so inadvertent double storage is prevented (although optionally possible for different conformations). This fact can even be used to find the name of a structure if only the structure is known (provided the compound is in the database).
Once in, the database will allow users to apply the various tools of the trade as one would expect from a modern chemical database - various search and retrieval methods are available, including unambiguous fragment and similarity search, stereoview display is possible and sorted lists can be generated. Indeed, ChemBrain can store any kind of data associated with a molecule. And, this is perhaps where the similarity with its counterparts on the market ends and the brains behind the package come to the fore.
ChemBrain uses the information associated with each molecule, whether in its built-in databases or imported in its artificial neural networks calculations. The package can learn from the stored data and allow one to predict properties of as yet unknown molecules.
The geometry-optimized 3D structures underpin these predictions as well as providing the basis for the classification, mapping, modelling, and selection of structures. Artificial neural networks are algorithms that mimic the neural connections in mammalian brain. They can thus associate any property to any other property, for instance, the geographical origin of a wine and its chemical constitution, molecular conformation and biological activity or toxicology. The networks are not static, however, and the more known associations that are fed to them the better their ability to find free associations for unknowns.
ChemBrain uses both the common neural network self-organising maps. The first is the Kohonen map, which consist of an input and an output layer, which is "trained" by forward-propagation unsupervised. The second, more flexible map is the back-propagation network, in which only the number of input and output neurons is defined by the task, and any number of hidden layers and neurons can be chosen freely. Training is achieved by a back-propagation procedure, which requires the properties of the input objects as a target in order to improve the connection weights, which means supervision is necessary. Thankfully, ChemBrain also has an algorithm that helps the user decide which strategy to adopt for particular tasks.
The creator of ChemBrain, Rudolf Naef of Swiss company ExpertSoft GmbH, suggests that the prediction of properties is one of the particular strengths of ChemBrain. The user has the option of selecting between several architectures of neural networks: mapping, modelling or classification. The program then searches its database for the most appropriate pre-calculated neural network and uses it for the prediction of the requested property or, if none is found, suggests training a new neural network based on molecules in the ChemBrain database structurally most similar to the query molecule.
Need to predict the solubility in water of your new drug lead. Simple, draw or import the molecule, click predict single value property and select water solubility and indeed, any of the other available properties, pKa for instance. You then select the neural network options, and on what property to base the training, mass, polarizability, or charge, say. You can limit the training to a group of types of molecule too, including most major drug classifications, from analgesic to peristaltic stimulant, agrochemicals, and even rodenticides. Get the settings right and out pops a list of molecules used in the training embedded within which is your drug molecule, with its solubility and pKa on display.
The same process can be used to determine any of numerous properties for almost any class of compound provided the training data are available. The reliability of a trained neural network can be tested in ChemBrain using its recall and prediction tests (where these are applicable). Need to know the melting point of a rodenticide or the bioavailability of an insecticide synergist? No problem.
Aside from its AI capacity, ChemBrain provides all the tools of the trade expected of a chemical database. But, it is the added value of the neural network algorithms that make it unique.
ChemBrain's sibling package, PiSystems augments the functionality of the chemical savvy database with a fast and reliable quantum chemistry calculation system. However, under Windows XP Pro, I found that I could only install PiSystems after I had uninstalled ChemBrain, which is rather inconvenient. To make sure both systems are installable I used a workaround that first installed PiSystems without the Borland Database Engine (BDE files), and then carried out a reinstallation of ChemBrain with the BDE. This worked fine in the end, but a warning within the setup would be useful to avoid such gremlins. I asked ExpertSoft about this and he says that installation should be possible as long as no application, e.g. ChemBrain itself or another application is running the BDE.
 Nevertheless, not only can you store, collate, search and retrieve molecules, as well as predicting the properties of unknowns, PiSystems allows you to generate their electronic spectra and work out their light absorption properties, i.e. the colour of organic molecules.
Nevertheless, not only can you store, collate, search and retrieve molecules, as well as predicting the properties of unknowns, PiSystems allows you to generate their electronic spectra and work out their light absorption properties, i.e. the colour of organic molecules.
PiSystems is a standalone system, but generation of new molecules is almost exactly the same as with ChemBrain. One can modify pre-generated fragments or alternatively draw them from scratch. Heteroatoms can be added, selected from a given set for which SCF-parameters are available. Experienced users may modify these parameters within limits which are controlled by the program. And, again, you can carry out a rapid optimisation of the geometry of the molecule to get a "clean" structure from which to start the interesting tasks - simulating electronic absorption spectra.
The calculated spectra are displayed in what ExpertSoft describes as a "close-to-reality fashion" by overlaying vibrational bands, their relative intensities being calculated by standard methods. The spectral range can be altered and even the direction of the spectrum changed as well as basic display parameters such as black on white or white on black display.
The program can then be used to provide insight into the dynamics of the electronic excitation within a molecule. For instance, a graphical display can be used to reveal the direction of the transition moment for the lowest electronic excitation of a molecule and its calculated intensity. Optionally, transition moments for the second and third electronic excitation can also be viewed.
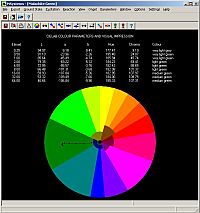
Concomitant with spectral prediction is the possibility of determining the colour of a conjugated molecule, such as a dye in solution. Standard CIELAB colour modelling methods are used to translate the calculated absorption spectrum into a simulated concentrations series in an inert media. The colour of the dye is then revealed in the software at different concentrations. This demonstration is, of course, more than a neat trick that would be useful in colour chemistry lessons even at high school level, it can be used by any chemist developing dyes for products as diverse as textiles to printer inks.
PiSystems goes much further than the basic prediction of electronic absorption spectra of conjugated molecules and the dynamic influences of the excitation within such molecules. It can be used to help in organic synthesis planning. If, for instance, a chemist wishes to create a new molecule with a shift in its long-wave absorption band, then PiSystems will allow the user to investigate the effects of adding a particular substituent at a certain point in the molecule. For example, substitution of a molecule with an amide group might shift the long-wave absorption band of a molecule towards shorter wavelengths if attached at certain points, while adding it to another centre would be revealed to shift the absorption to the other end of the spectrum.
It is also possible to extend this capability to investigating reactivity related characteristics. If the most reactive centre within a conjugated molecule is of interest then it is possible to determine where it would be with reference to reactive nucleophilic or electrophilic reagents, on the basis of its electronic excitation profile.
System requirements - any PC system running Windows95 onwards. However, installation on a minimal Pentium Pro system running Windows98 is impossibly slow. On a mid-range PC (256Mb RAM, 1.4 MHz CPU), installation and operation are fine. 10MByte of free storage space is needed for each not including the database, which is requires about 10-20 kBytes per molecule for PiSystems and a minimum of 8MByte space for ChemBrain.
ChemBrain 2002 version 2.4 and PiSystems 2002 version 5.4 are available for 30-day trial download from www.expertsoft.ch/sciencetent is pending on the device and the team is now seeking a commercial
partner.